Amphibian-like ranaviruses are pathogens of amphibians, reptiles and fish. They are capable of causing a severe disease shown to have resulted in the collapse of amphibian communities in one region. Ranaviruses have double-stranded DNA genomes which encode around 100 genes. Their large genomes and particle sizes are characteristics shared by other viruses in a group called the “large DNA viruses”.
This “large DNA virus” group, though it includes some other horrific pathogens such as smallpox, has often been viewed as a bit dull: somewhat evolutionary inactive – by virtue of (usually assumed) low substitution rates and speciation alongside hosts, and somehow less threatening than other groups of viruses with RNA genomes that can evolve very rapidly over the course of an epidemic.
There’s been a fair bit of work over the last decade though that shows the genomes of large DNA viruses are home to all sorts of exciting processes. They seem to be great at mopping up bits of the genomes of other organisms, they can do sex a lot, and their substitution rates can be as high as very sloppy RNA viruses.
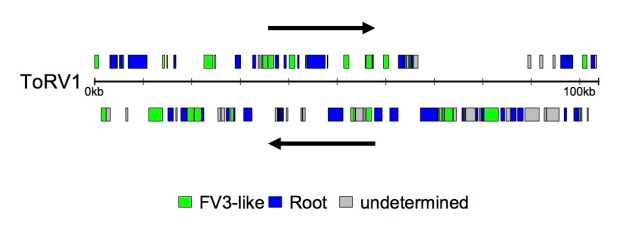
I’ve just published some analyses of genomic variation and evolutionary processes in the amphibian-like ranaviruses. I took all the publicly available genomes at the time and had a look at how genome content varied across the group – what’s shared and what’s not? I also wanted to know who was related to who in this group and the robust ways to assess this. Finally I wanted to know which genes had been subject to dynamic processes like recombination and positive selection during their evolution. These questions are interesting because these viruses can be important pathogens and genomic variation is likely to yield variation in key pathogen traits such as virulence and host range.
Key findings:
- Ranavirus genomes have been subject to dynamic genome change which may be linked to important aspects of their biology.
- Ranavirus genome content varies systematically and individually – there are small groups of genes which appear specific to major clades or that are lost in one such clade
- Recombination and positive selection are widespread in amphibian-like ranavirus genomes:
- some genes with evidence of recombination have previously been used for reconstruction of phylogenetic relationships but failure to account for recombination might lead to erroneous inferences
- of the 16 viruses studied, positive selection along terminal branches was found most frequently in the spanish CMTV which has had a major impact on amphibian communities in the Picos de Europa
- some viruses from aquaculture facilities also have a relatively high number of genes under positive selection. If such selection is for greater virulence (likely given the availability of susceptible hosts at high stocking densities and subject to ongoing replenishment) then spill back of these viruses into the wild could have important implications for conservation of herpetofauna.
- Recombination among divergent viruses might have given birth to a mosaic virus infecting a captive reptile, exposing the possibility that trade in herpetofauna might increase opportunities for such interactions between viruses previously separated geographically.
- We don’t have functional information for the majority of ranavirus genes and this hampers our ability to interpret variation. Gene knockout strategies offer potential in trying to explore variation in genome content.
Download the paper here
Citation: Price SJ (2016) Comparative genomics of amphibian-like ranaviruses, nucleocytoplasmic large DNA viruses of poikilotherms. Evolutionary Bioinformatics 2015:Suppl. 2 71-82 (published 25/10/16) DOI: 10.4137/EBO.S33490.
